Protein Phylogeny
Phylogenetic Trees and Clustal Omega
Phylogenetic trees are used a means to visualize the evolutionary history of an organism or, in the case of this project, a protein. The number of branches that are between two species indicates their relatedness, with more branches indicating that the species are distantly related. Phylogenetic trees were created for the HFE protein using Clustal Omega, a sequence alignment bioinformatic tool sponsored by the European Bioinformatics Institute. In this program two types of trees can be made. One tree uses the neighbor joining method in which creates an unrooted tree based off of evolutionary distance data [1]. The other type of tree uses the average distance method in which the percent of sequence identity is used to calculate branch distances.
In addition, two different types of matrices can be used to generate each tree. The BLOSUM62 method scores the likelihood that one amino acid would be substituted for another. On the other hand Percent Identity scores how many identical amino acids there are between sequences to determine similarity. Phylogenetic trees using each of these methods are shown in the figures below.
In addition, two different types of matrices can be used to generate each tree. The BLOSUM62 method scores the likelihood that one amino acid would be substituted for another. On the other hand Percent Identity scores how many identical amino acids there are between sequences to determine similarity. Phylogenetic trees using each of these methods are shown in the figures below.
Phylogenetic Trees for HFE Homologues
Below are four phylogenetic trees for homologues of human HFE that were created using the algorithms described previously. To enlarge one of the trees simply click on the picture.
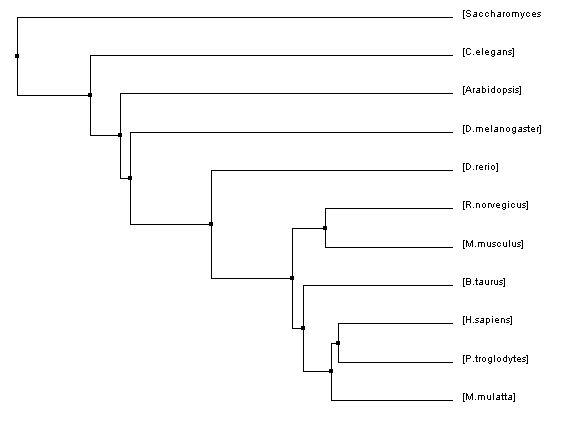
Figure 1: Average distance tree using BLOSUM62
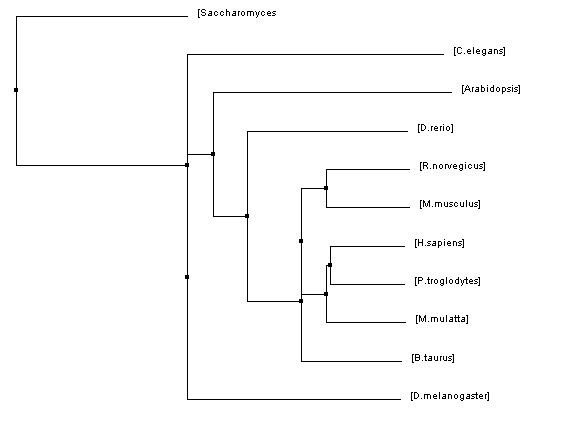
Figure 3: Neighbor joining tree using BLOSUM62
|
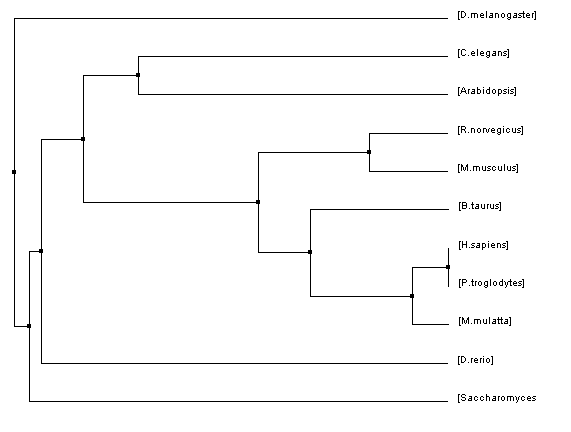
Figure 2: Average distance tree using percent identity
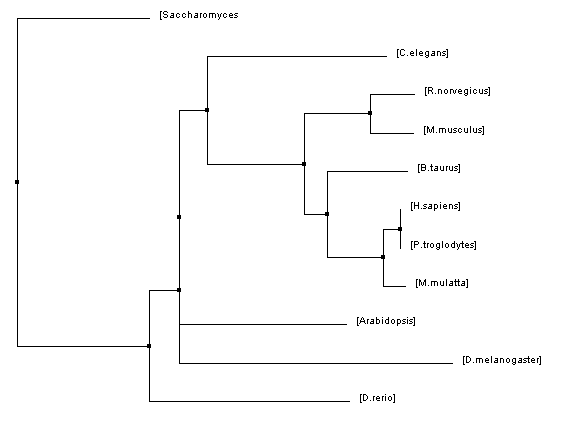
Figure 4: Neighbor joining tree using percent identity
|
Analysis
The phylogenetic trees that were created using all of these algorithms were similar in many regards but they also all had their fair share of differences. Homo sapiens and Pan troglodytes were within the same clade on every tree. This intuitively makes sense as chimpanzees are considered to be human's closet ancestors and the sequences for the HFE protein in both species is identical. From there, every step out from the humans (indicating decreasing similarity/more and more distant relations) was a vertebrate ending just before D. rerio. This also makes sense as vertebrates have blood and hemochromatosis can be considered a blood disorder.
However, once D. rerio was reached there was some variability on these trees. One would think that D. rerio would be grouped with all of the vertebrates (such as in Figure 1) because it is a vertebrate as well. However, in some of the trees species such as Arabidopsis and C. elegans were shown to be more closely related to humans than D. rerio. Given the variety of different algorithms that were used to make these trees it is unsurprising that there would be some variability like this. The fact that D. rerio was not always grouped with the vertebrates could have a significant meaning or it could just be due to chance because of the variability of the algorithms.
One interesting thing that I would like to draw your attention to is in Figure 1. In this tree, all of the vertebrates are grouped together all the way through D. rerio. Then, the next most closely related species to human, according to this algorithm, is D. melanogaster. I feel that it makes sense for the fruit fly to be the next branch on the tree because they possess blood-like cells called hemocytes [2].
However, once D. rerio was reached there was some variability on these trees. One would think that D. rerio would be grouped with all of the vertebrates (such as in Figure 1) because it is a vertebrate as well. However, in some of the trees species such as Arabidopsis and C. elegans were shown to be more closely related to humans than D. rerio. Given the variety of different algorithms that were used to make these trees it is unsurprising that there would be some variability like this. The fact that D. rerio was not always grouped with the vertebrates could have a significant meaning or it could just be due to chance because of the variability of the algorithms.
One interesting thing that I would like to draw your attention to is in Figure 1. In this tree, all of the vertebrates are grouped together all the way through D. rerio. Then, the next most closely related species to human, according to this algorithm, is D. melanogaster. I feel that it makes sense for the fruit fly to be the next branch on the tree because they possess blood-like cells called hemocytes [2].
References
1. Saitou, N. and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4): 406-425.
2. Fruit Fly Will Aid In Blood Studies: http://www.hhmi.org/news/fruit-fly-will-aid-blood-studies
3. Clustal Omega: https://www.ebi.ac.uk/Tools/msa/clustalo/
2. Fruit Fly Will Aid In Blood Studies: http://www.hhmi.org/news/fruit-fly-will-aid-blood-studies
3. Clustal Omega: https://www.ebi.ac.uk/Tools/msa/clustalo/
